摘 要 以异烟酸-巴比妥酸作为显色剂代替异烟酸-吡唑啉酮测定杏仁油中的氰化物。通过实验,校正曲线在0~2μg范围内,线性关系良好,回归方程Y=0.4937X-0.OO05,相关系数r=0.9991,最低检测浓度为0.1mg/kg,加标回收率为92.4%。该方法克服了苯甲醛与吡唑啉酮反应产生白色混浊的干扰,可用于杏仁油中氰化物的测定。
关键词 氰化物 杏仁油 测定
山杏仁中含有氰化物,成人致死量平均50粒。在制取杏仁油过程中,氰化物不可避免地进入杏仁油中。因此杏仁油生产过程中,比较关键的一环是去除氰化物,使其在安全范围内。目前,关于杏仁油提取和脂肪酸组成方面的研究较多,但还没有建立杏仁油国家标准或行业标准,对杏仁油中氰化物含量的研究也未见报道。氰化物的检测方法在GB/T5009.48-2003《蒸馏酒及配置酒卫生标准的分析方法》和GB/T5009.36-2003《粮食卫生标准的分析方法》中均采用异烟酸-吡唑啉酮分光光度法[1,2]。笔者在实际测量杏仁油中氰化物时发现,在加入显色剂后出现不同程度的白色混浊,严重干扰测定效果。参考GB/T5750.5-2006《生活饮用水标准检验方法无机非金属指标》氰化物测定方法第二法,笔者将显色剂更换为异烟酸-巴比妥酸,消除了混浊的干扰[3]。
1 材料与方法
1.1 原理
试样中氰化物经水蒸气蒸馏后被碱溶液吸收,与氯胺T生成氯化氰,然后与异烟酸-巴比妥酸反应生成紫蓝色化合物,于600nm波长处比色定量。
1.2 仪器
水蒸汽蒸馏装置;25ml具塞比色管;721型分光光度计。
1.3 试剂
氰化物标准使用液(1μg/ml)、酒石酸、100g/L乙酸锌溶液、40g/L氢氧化钠溶液、12g/L氢氧化钠溶液、乙酸溶液(3+97)、lg/L酚酞指示剂、136g/L磷酸二氢钾溶液、10g/L氯胺T溶液(现用现配)。
异烟酸—巴比妥酸溶液:称取2.0g异烟酸和1.0g巴比妥酸,溶于60~70℃的12g/L氢氧化钠溶液100ml中。
以上试剂均为分析纯,参照GB/T 5750.5-2006 《生活饮用水标准检验方法无机非金属指标》进行配制。
1.4 方法
直接称量杏仁油试样10g于蒸馏瓶中,向其中加入100g/L乙酸锌溶液20ml,再加入酒石酸1~2g,立即通入水蒸气进行蒸馏,用装有10ml40g/L氢氧化钠溶液的100ml容量瓶,收集馏液近100ml,取下容量瓶,加水至刻度。
吸取l0ml蒸馏吸收液,置于25ml具塞比色管中。另取25ml具塞比色管10支,分别加入氰化物标准使用液0ml、0.10ml、0.20ml、0.40ml、0.60ml、0.80ml、1.00ml、1.50ml、2.00ml、2.50ml,各加12g/L氢氧化钠溶液至l0ml。向试样及标准管中各加l滴酚酞指示剂,用乙酸溶液调至红色刚好消失。向各管中各加入3ml磷酸二氢钾溶液和0.25ml氯胺T溶液,混匀。放置2min后,向各管中加入5ml异烟酸-巴比妥酸溶液。25℃下显色15min,于波长600nm下,用3cm比色皿,以零管调节零点,测定试样和标准系列溶液的吸光度。绘制校正曲线,在曲线上查出试样管中氰化物的含量。
计算公式
A×100×1000
P (mg/L)=
m×10×1000
A—试样氰化物含量(μg)
m—试样质量(g)
2 结果
2.1 校正曲线和回归方程
校正曲线在0~2μg范围内,线性关系良好,回归方程Y=0.4937X-0.OO05,相关系数r=0.9991。最低检测量为0.1μg,若取10g试样测定,最低检测浓度为0.1mg/kg。
2.2 方法的精密度和准确度试验
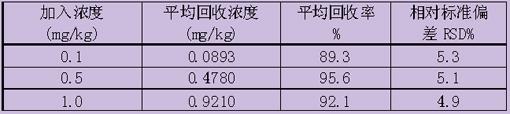
分别做0.1、0.5、1.0 mg/L3种浓度的加标回收试验,每种浓度平行测定5次,结果见表l。
表1. 精密度和准确度测定结果
3 讨论
3.1杏仁油中含有苯甲醛,苯甲醛又名苦杏仁油,普通品为无色至淡黄色液体,沸点179℃,有苦杏仁味,微溶于水,能与水蒸气一同挥发。正是因为它能与水蒸气一同挥发的特性,在试样处理时,无论采用直接蒸馏法还是水蒸气蒸馏法都无法消除干扰。另外杏仁中苦杏仁苷是由戊胆二糖、氢氰酸、苯甲醛组成,它在水解产生氢氰酸的同时,也产生苯甲醛。实验证明,只有苯甲醛与吡唑啉酮产生白色浑浊,而且浑浊只在中性和酸性条件下产生。异烟酸-吡唑啉酮是测定氰化物的最终显色剂,苯甲醛与吡唑啉酮产生白色浑浊时,一方面溶液浑浊影响比色,另一方面也影响了氯化氰与异烟酸-吡唑啉酮生成蓝色,因此异烟酸一吡唑啉酮分光光度法不适用于测定杏仁油中氰化物。测定氰化物的化学方法现在常用的主要有异烟酸-吡唑啉酮法、砒啶-吡唑啉酮法、异烟酸-巴比妥酸法等。吡唑啉酮用巴比妥酸替换,取得满意的实验结果,显色时间也由40min缩短到15min。这在徐向阳等的《杏仁露氰化物测定方法的研究》中有过报道[4]。
3.2当氰化物以HCN存在时易挥发,从用乙酸溶液调完pH值后,每一步都要迅速操作,并随时盖好塞子。
3.3氯胺T含量的改变,常常是导致实验失败的的原因,氯胺T易受阳光和空气作用而逐渐分解失效,因此必须储存在密封的容器里,并放在阴凉干燥处,在使用前现配,当配置后出现混浊沉淀物时,不宜再用。在显色阶段,用氯胺T氧化为氯化氰的过程,由于氯化氰的沸点很低(13℃),所以混匀动作不宜过猛。
3.4蒸馏操作过程中,控制蒸馏速度为2~4ml/min,过快易使氰化物损失,同时应控制好吸收液的碱浓度,冷凝管的下端要插入吸收液的液面下,使吸收完全。馏出液在蒸馏完后不能呈酸性,可在吸收液中加2滴酚酞指示剂,使其在吸收完后依然保持红色。而在GB/T5009.36中以10g/L的氢氧化钠溶液作吸收液,满足不了要求,应提高吸收液碱含量或增加吸收液用量。
(本文实验过程中得到朝阳市疾病预防控制中心徐向阳的大力帮助,同时也得到了朝阳市产品质量监督检验所李玉红和武冬梅的帮助,在此一并表示感谢!)
参考文献
[1] GB/T 5009.36-2003,粮食卫生标准的分析方法[S].
[2] GB/T 5009.48-2003,蒸馏酒及配制酒卫生标准的分析方法[S].
[3] GB/T 5750.5-2006,生活饮用水标准检验方法无机非金属指标[S].
[4]徐向阳,翟凤兰.杏仁露中氰化物测定方法的研究[J].中国食品卫生杂志,2002,14(6):20-22.
(作者单位:辽宁省朝阳市产品质量监督检验所 辽宁省北票市卫生防疫站)